An der Medizinischen Universität Innsbruck wurde unter der Leitung von PD Dr. Ines Kapferer-Seebacher (Parodontologin) und Univ. Prof. Dr. Johannes Zschocke (Humangenetiker) in einer internationalen Kooperation die genetische Ursache des parodontalen Ehlers-Danlos Syndroms (pEDS) identifiziert und die Erkrankung zum ersten Mal systematisch klinisch charakterisiert. Beide waren auch an der Erstellung der derzeit gültigen Klassifikation von 2017 beteiligt.
Die Ehlers-Danlos Syndrome sind eine heterogene Gruppe von dreizehn angeborenen Bindegewebserkrankungen, und das pEDS ist eine davon. Die Existenz des pEDS als spezifische Entität wurde in den letzten Jahrzehnten immer wieder angezweifelt, da es anderen EDS Subtypen ähnelt.
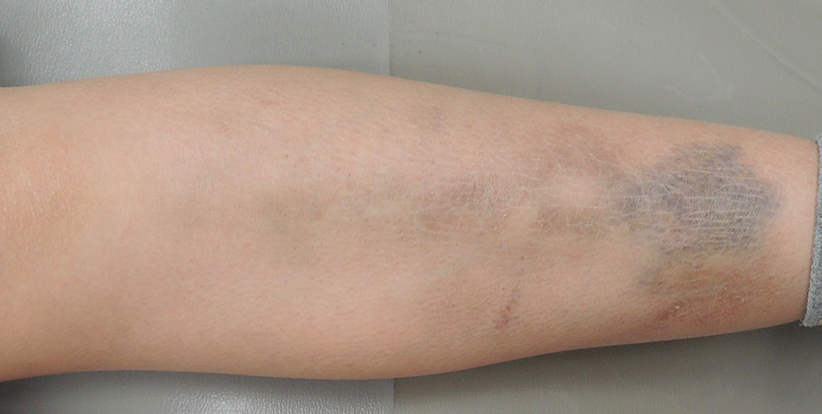
Typisches Merkmal des pEDS ist allerdings – im Gegensatz zu den anderen EDS Subtypen – eine rasch fortschreitende Destruktion des Parodonts im Jugend- oder Kindesalter. Hinzu kommen bräunlich atrophische Vernarbungen an den Schienbeinen und eine generelle Bindegewebsschwäche. Die Bindegewebsschwäche kann in manchen Individuen nur sehr milde ausgeprägt sein, sodass zum Beispiel nur die Fingergelenke überdehnbar sind. In anderen Individuen wiederum kann die Bindegewebsschwäche zu Aneurysmen und Organrissen führen. Bei wieder anderen Individuen zeigen sich rezidivierende Infektionen oder über Monate schlecht heilende Wunden. Dem autosomal-dominant vererbten pEDS liegt eine Mutation im Komplement 1 zugrunde, die von Kapferer-Seebacher et al. beschrieben wurde.
Damit stellt das pEDS einen bisher unbekannten Link zwischen dem klassischen Komplement-Aktivierungsweg und dem Bindegewebe dar, der im Rahmen eines FWF – Projektes nun an der Medizinischen Universität Innsbruck weiter untersucht wird.
Beim „American College of Medical Genetics and Genomics Meeting 2018“ wird das pEDS vom Innsbrucker Team präsentiert werden.
Fotos © Ines Kapferer-Seebacher